ABSTRACT
DNA methylation is one of the major epigenetic mechanisms that play an important role in a variety of cancers. Several high throughput profiling techniques (MeDIP-seq, MIRA-seq, MBD-seq, MethylCap-seq, MethylC-seq, BS-seq) have been developed to study genome-wide methylation patterns. Affinity-based enrichment of methylated DNA sequences with methyl-CpG binding domain proteins followed by next generation sequencing (MBD-seq) utilizing the MethylMiner™ Methylated DNA Enrichment kit has been shown to be a powerful alternative to MeDIP-seq and the whole methylome sequencing technology of BS-seq. Several recent studies applied methods based on tags density or tags count normalized by the CpG density. Similar to many of the peak detection programs, low resolution is the major disadvantage of using tags density based methods for MBD-seq data analysis. In this study, we performed high sequencing depth MBD-seq in the human breast-cancer MCF-7 cell line. The result shows that with ~100 million unique mapped tags (approximately five lanes using a GAII sequencer) from 500mM and 1000mM elutes the coverage of the MBD-seq data become close to a saturation point. A bi-asymmetric-Laplace model (BALM) was developed to analyze MBD-seq. We compared the resolution of BALM to that of several ChIP-seq analysis tools. The results demonstrate the program’s superior ability to distinguish methylation statuses of closely positioned CpG sites. This study demonstrates that MBD-seq combined with the new program is potentially a powerful tool to capture genome-wide DNA methylation profiles with high efficiency and resolution.
Please cite the BALM paper if you use this tool. Download the paper here.
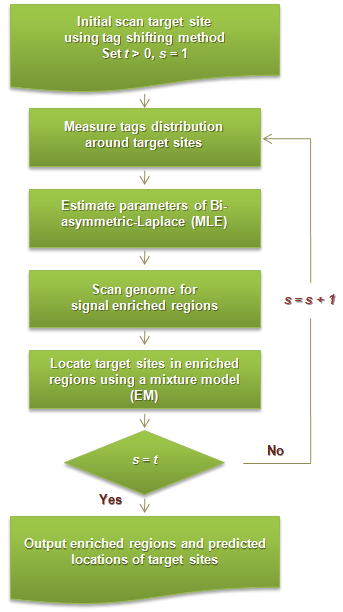
BALM-ALGORITHM