Introduction
HiSIF is a novel computational software tool on Hi-C data analysis, including a Poisson Mixture Model, with an Expectation Maximization algorithm (EM) followed by a power-law decay background model to filter background-ligation events.
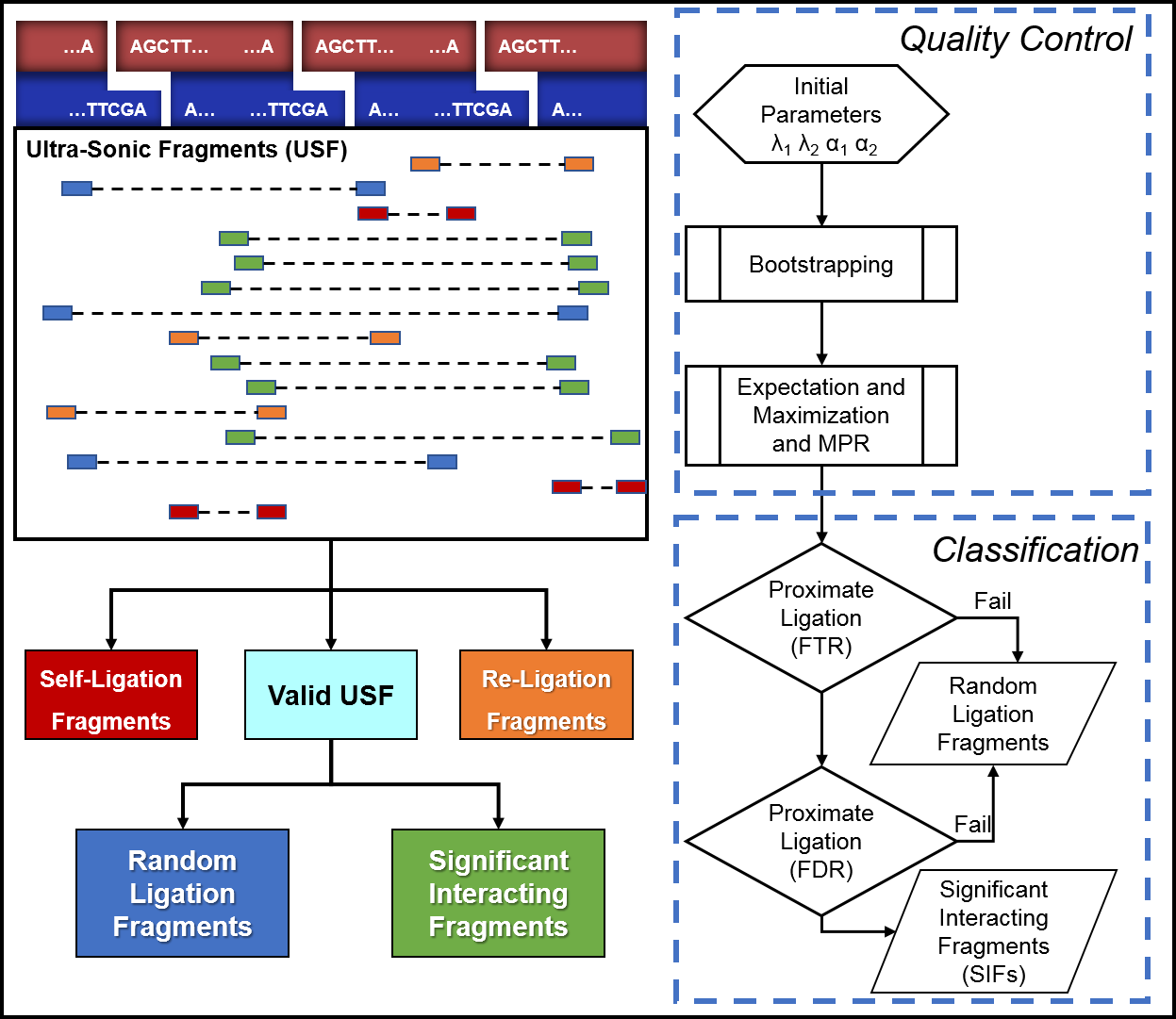
HiSIF is composed by two main modules: Quality Control and Classification.
Quality Control
• initialize the parameters
• adjust parameters to the dataset
• characterizes different interactions: self-ligation event, re-ligation event and valid-ligation event
Classification
• apply Fragment Threshold and log likelihood (LLH) of power law filter
• classify valid USF as: random ligation event and proximate ligation event
The detailed description can be found in our manuscript.
Download
You can download HiSIF by following link and current version is v1.0.
http://jinlab.net/HiSIF/HiSIF_V1.00.zip
HiSIF will be maintained on Github, please find the latest HiSIF on :
Installation
HiSIF was implemented by using C++/C with parallel processing being written in C. It has been compiled and run exclusively on Linux operating systems. This tool only requires the g++ compiler and a reference genome for HG19. Standalone CERN ROOT C++ framework is used to extract fit parameters of the CTS interactions. A small C tool is provided to process the initial data from NCBI. HiSIF supports all of the available three Hi-C protocols (Hi-C, TCC and in situ Hi-C). The tool is named from the fact that it is designed to find the significant interactions from a given sample of Hi-C reads.
System Requirements:
•Operating System: Linux/Unix
•CPU Cores: at least 32
•RAM: 32GB minimum (will not be able to run in-situ)
•Hard Disk Space: >100GB (more as datasets get larger)
You can install it by following commands:
unzip HiSIF_V1.00.zip
cd HiSIF_V1.00
make
This will create 2 executables in the bin directory:
1) proc - used for preprocessing of data
2) HiSIF - binary for algorithm
Pipeline Overview
1. Map using bowtie2 or similar software
2. convert the BAM file format to a 6-column file format
3. use 'split -l' to split the produced file into many for parallelization
4. use the 'proc' tool to sort these text files, producing a directory of chr files
5. use HiSIF with specific parameters
Quick Start
After configuration file config_hisif.txt is changed as necessary, perform the following:
chmod 755 runhisif.sh
./runhisif.sh config_hisif.txt
Configuration file config_hisif.txt including:
SAMTOOLS: path for samtools
HISIF: path for HiSIF
HICPROBAMPATH: the bam file path from HiC-Pro output, leave it empty when HiC-Pro output is not used.
BAMPATH: the path with multiple bam files, leave it empty when not multiple bam files
BAMFILE: the path and file name of one bam file
OUTPUT: the path for saving the HiSIF output
SAMPLE: the name of the sample
PREPROCESSING: "True" need preprocessing, "False" doesn't need preprocessing
REFERENCE_GENOME: the path for reference genome
CUTTING_FRAGMENTS: the genome enzyme cutting sites fragments
READ_LENGTH: sequencing reading length
CUTTING_SITE_EXTENT: the extent of cutting site, default 500
BIN_SIZE: bin size, normally HindIII is 20000, MboI is 3000
FRAGMENT_SIZE: Output fragment size, normally is the resolution
THRESHOLD_FIRST: first value of SIF threshold value, default is 1
THRESHOLD_LAST: last value of SIF threshold value, default 5
PERCENTAGE: Percentage of dataset for bootstrapping
POISSON_MIXTURE1: Poisson mixture model parameter 1, default 1
POISSON_MIXTURE2: Poisson mixture model parameter 2, default 29
ITERATIONS: Bootstrapping iterations, default 2
CHILD_PROCESSES: Limit number of child processes used, default 0 (no limt)
LOGFILE: the log file name, default runhisif.log
Customized Running
The running could be customized by user referring to the provided Linux shell file runhisif.sh
Or refer to the following introduction.
There are two steps to use HiSIF: pre-processing, and running HiSIF.
Step1: Pre-Processing
This method assumes mapping with bowtie2 or a similar tool has been done.
Using the SAM/BAM files from the mapping, transfer them to 6-column text file:
chr1 pos1 strand1 chr2 pos2 strand2
Please note strand is 1 for positive strand and 0 for negative strand. Each chromosome need only the number and chrX is 23 and chrY is 24. That means chr1/chr2 must be 1-24.
Note that with very large output files, the 'proc' executble will fork for each file. It is suggested to split this file into 10 files, to speedup pre-processing.
Creating the chr-by-chr files Before using the 'proc' program, it is suggested to split up the text file into many different files (at least 10), using 'split -l'. The file prefixes when finished are: chr1.tmp chr2.tmp ... chrX.tmp Usage: proc: <-r or -t> -t traditional 6-column file format -r rao format Note: for some in-situ datasets, they produce a different formatting. Please refer to the FORMATS file for more information, and look at how the python script is used.
Step2: Running HiSIF
Program: HiSIF - HiC Significant Interaction Fragments Version: 1.0.0 HiSIF [options]
-g <DIR> reference genome directory
-c <FILE> cutting sites map .bed file
-p <INT> <INT> poisson mixture model parameters
-w <INT> <INT> <INT> readLength, cuttingSiteExtent, binSize
-t <INT> peakThreshold value, 1, 1.5, 2 and so on
-s <0.0-1.0> percentage of dataset for bootstrapping, default is 1
-i <INT> bootstrapping iterations, default is 50
-f <INT> output fragment size, default is the same as binSize
-x limit number of child processes used, default is 0 (no limit)
For example:
Run the following for HindIII digested Hi-C experiments
bin/HiSIF -g <hg19genome> -c <resources/hg19.HindIII.bed> -p 1 29 -w 50 500 20000 -t 1 -i 2 chrfiles
Run the following for MboI digested Hi-C experiments
bin/HiSIF -g <hg19genome> -c <resources/hg19.MboI.bed> -p 1 29 -w 50 500 3000 -t 1 -i 2 chrfiles
Results
All results are prefixed with (SAMPLE)t(THRESHOLD), threshold could be 1, 1.5, 2, 2.5, 3 and so on.
(SAMPLE)_t(THRESHOLD)_peak.txt: the significant interaction fragments with columns: chr1 start1 end1 chr2 start2 end2 counts FDR This is the major result, normally users only need this result for the subsequent analysis. (SAMPLE)_t(THRESHOLD)_BootStrapping.txt: system parameters for boot strapping (SAMPLE)_t(THRESHOLD)_maxIteration.txt: system parameters for iteration (SAMPLE)_t(THRESHOLD)_PerChr.txt: number of enzyme cutting fragment for each chromosome (SAMPLE)_t(THRESHOLD)_randomDis.txt: random distribution of sequencing reads (SAMPLE)_t(THRESHOLD)_randomDisProb.txt: random distribution probability of sequencing reads
Troubleshooting
1. When proc is running, there is maybe an error: Error readInteractingRegions:: read error. >>>: Is a directory This is the system reminding of reading the current directory, which does not affect successfully performing. 2. When HiSIF is running, there are maybe some errors like: Error: could not read the first line : Is a directory This is the system reminding of reading some sub-directories in the reference genome, which does not affect successfully performing. 3. Error of "Segmentation fault": IF too many reads overburden the memory, the segmentation fault error will occur. Please run HiSIF with chromosomes individually to reduce the memory requirements.