Introduction¶
ChIP-exo is a new emerging novel protocol in which a lambda exonuclease is introduced into the ChIP system (see <https://en.wikipedia.org/wiki/ChIP-exo>). This exonuclease degrades unbound double-stranded DNA in 5′-3′ direction, to within a few nucleotides of the protein binding site, then the exonuclease-treated 5′-ends (or exo-5’-ends) are subjective to sequencing, and high concentration of exo-5’-ends at one location would represent a protein protected boundary that usually termed as “border” in ChIP-exo analysis. This new technique greatly increases the resolution of binding sites to single base pair. Here, in our previous studies, we have modified the protocol as ChIP-ePENS (ChIP-exo paired-ends sequencing, see the following figure), in which we not only collects exonuclease-treated 5’-end (exo-5’-end) but also sonicated 3’-end (son-3’-end) together.
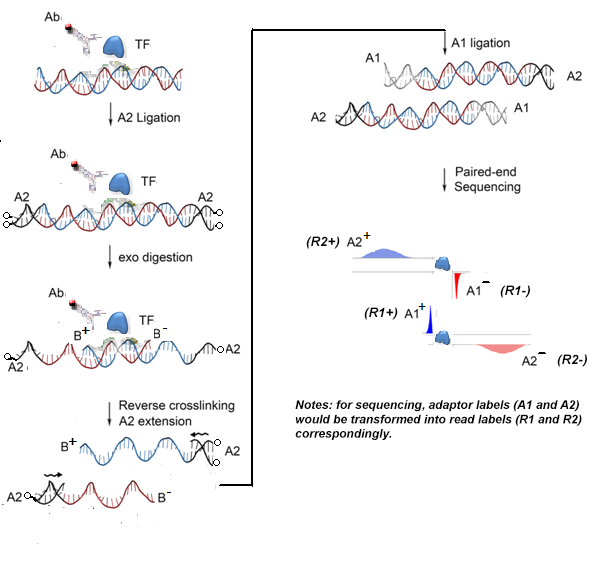
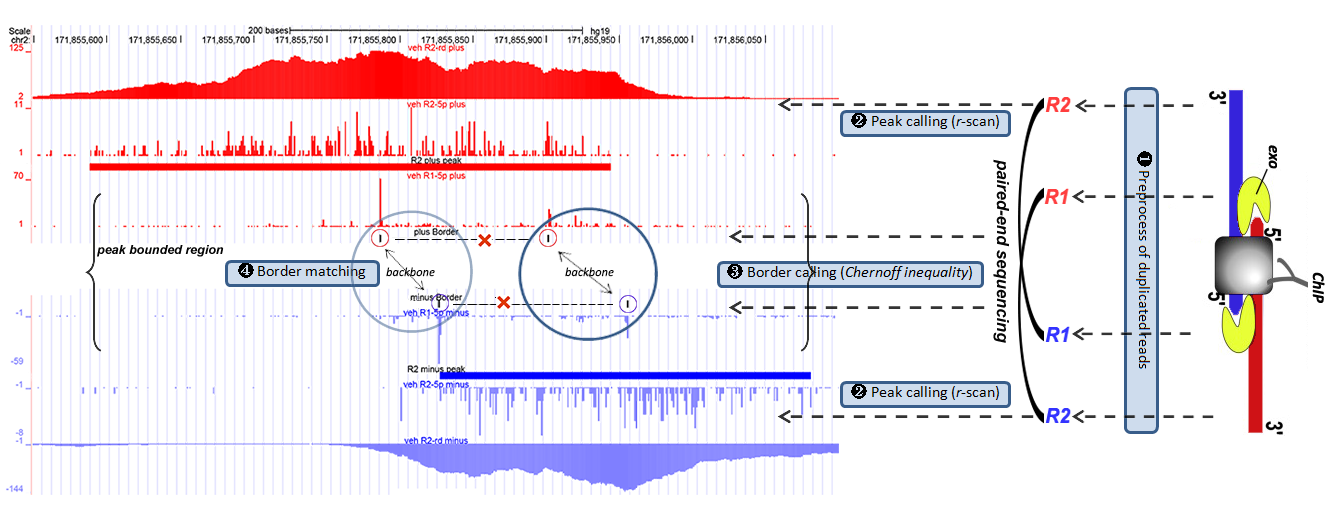
For the above protocol, we have developed a novel algorithm, ePEST (ChIP-exo paired-end sequencing processing toolkit), which leverages on the statistical powerful of r-scan for detecting binding peaks with son-3’-end reads and Chernoff inequality for identifying precise borders with exo-5′-end reads, respectively. The detected borders are further modelled as graphical components and classified into distinct border patterns based on their orientation and spacing. Our novel computational algorithm for ChIP-ePEST data analysis is illustrated in the following figure, and is composed of four steps.
(1): Preprocess of raw and mapped reads.
(2): r-scan for calling peaks on R2 reads.
(3): Inequality of Chernoff bound for calling borders on R1 reads.
(4): An iterative outlier-cutting strategy for border-matching in a graphical model.
The detailed description can be found in our manuscript.
Installation¶
The software is implemented by Python(>=3.3), and several third-party libraries are required:
cement (>= 2.0.2) http://cement.readthedocs.org/
networkx (>= 1.8.1) https://networkx.github.io/
numpy (>=1.8.0) http://www.numpy.org/
scipy (>=0.13.2) http://www.scipy.org/
pysam (>=0.7.7) https://github.com/pysam-developers/pysam
treelib (>=1.2.7) https://github.com/caesar0301/treelib
Usage¶
To use the software, it’s simply to unzip the downloaded file into a directory, and install the above required libraries, then run the following command to get more information about usage:
python ePEST.py --help
- usage: ePEST.py [-h] [–debug] [–quiet] [-o OUTPUT] [-r REGEX] [-p float]
- [-D {True,False}] [-d int] [-q float] [-R int] [-s int] [-S int] [-c float] [-k float] [-t int] –input.bam
ePEST: ChIP-exo paired-end sequencing processing toolkit, for peak-calling and border identify.
- positional arguments:
- –input.bam the bamfile from ChIP-exo. REQUIRED
- optional arguments:
-h, --help show this help message and exit --debug toggle debug output --quiet suppress all output -o OUTPUT, --output OUTPUT the output fold for results. Default is the current directory. -r REGEX, --regex REGEX the read id pattern. The Default is setted with Solexa platform: [a-zA-Z0-9]+:[0-9]:([0-9]+):([0-9]+):([0-9]+).* -p float, --pvalue float the p value of statistical significant for trigging peak-calling. Default=1e-5. - -D {True,False}, –dedup {True,False}
- the flag for removing those duplicated reads; only works for paired sequencing. Default=True.
-d int, --dist int the cut-off distance between reads touched on the flow chip for optical duplicate detection. Default=100. -q float, --qvalue float the pvalue used for amplification fragments detection. Default=0.001. -R int, --rscan int the r-scan parameter, a minimal number of R. Default=20. -s int, --minstep int the minimal number of base pairs for r-window spanning. Default=1. -S int, --maxstep int the maximal number of base pairs for r-window spanning. Default=8. -c float, --chernoff float the probability of chernoff inequality. Default=0.05. -k float, --outlier float the kth-fold std deviation for outlier detection. Default=2.0. -t int, --threads int the number of threads enabled for parallel computation.
Walkthrough example using ChIP-ePENS FoxA1 data in vehicle LNCaP¶
The following commands have been tested on a server with 64 CPUs/256G Memory, CentOS release 6.3.
Step1: Download alignment data¶
Download FoxA1 ChIP-ePENS data from our website (<http://jinlab.net/ePEST/FoxA1/vehLNCaP_FoxA1_ChipEXO.R1R2.Paired.Align.bam>)
wget http://jinlab.net/ePEST/FoxA1/vehLNCaP_FoxA1_ChipEXO.R1R2.Paired.Align.bam
This BAM files has been aligned to hg19 by bowtie1 and preprocessed by samtools, only unique mapped read-pairs are remained for further analysis.
bowtie -v 3 -k 2 -m 1 -p 15 --fr -I 20 -X 400 -S /data/yez/resource/hg19/hg19 -1 /data/yez/Projects/ChIP-exo/FOXA1_veh_ChipEXO_NoIndex_L005_R1.fastq -2 /data/yez/Projects/ChIP-exo/FOXA1_veh_ChipEXO_NoIndex_L005_R2.fastq /data/yez/Projects/ChIP-exo/vehLNCaP_FoxA1_ChipEXO.R1R2.Paired.sam
Step2: Running ePEST pipeline¶
Be sure of successful installation of Python3 and extra libraries before running the ePEST.
python ePEST.py -D True -p 1e-8 -R 25 -t 12 -c 0.05 -k 2.0 -o ePEST_vehFOXA1 /data/yez/Projects/ChIP-exo/vehLNCaP_FoxA1_ChipEXO.R1R2.Paired.Align.bam
Several output files will be deposited into the fold ePEST_vehFOXA1. For example, under the “Peak” fold, there are paired-peaks BED files on each chromosome. And under the “Border” fold, two kinds of files can be found, one is the BED file for each individual border information, the other is the json file for further graph-based analysis. Especially, for each row in the border BED file, those columns are interpreted as following:
chrid start end bordername depth strand chernoff peakid compid pnb
noted: compid means this border belongs to this graph-component, and pnb means the numbers of plus borders, minus borders and backbones in this component.
Step3: Visualization of the output from ePEST pipeline¶
The peak and border BED tracks produced by ePEST can be further processed for visualization in the UCSC genome browser by setting them up.
track type=bigWig name="veh R2-rd plus" description="veh R2-rd plus" visibility="full" color=255,0,0 bigDataUrl=http://jinlab.net/ePEST/FoxA1/vehLNCaP_FoxA1.ChipEXO.R1R2.Paired.Align_R2_plus.sorted.bw
track type=bigWig name="veh R1-5p plus" description="veh R1-5p plus" visibility="full" color=255,0,0 bigDataUrl=http://jinlab.net/ePEST/FoxA1/vehLNCaP_FoxA1.ChipEXO.R1R2.Paired.Align_R1_plus.5end.bw
track type=bigBed name="FOXA1 Peak Pairs" visibility=2 itemRgb="On" db=hg19 bigDataUrl=http://jinlab.net/ePEST/FoxA1/ePEST_vehLNCaP_FoxA1_peakcall.paired.sorted.bb
track type=bigBed name="FOXA1 Borders" visibility=2 db=hg19 bigDataUrl=http://jinlab.net/ePEST/FoxA1/ePEST_vehLNCaP_FoxA1_borders.sorted.bb
track type=bigWig name="veh R1-5p minus" description="veh R1-5p minus" visibility="full" color=0,0,255 bigDataUrl=http://jinlab.net/ePEST/FoxA1/vehLNCaP_FoxA1.ChipEXO.R1R2.Paired.Align_R1_minus.5end.bw
track type=bigWig name="veh R2-rd minus" description="veh R2-rd minus" visibility="full" color=0,0,255 bigDataUrl=http://jinlab.net/ePEST/FoxA1/vehLNCaP_FoxA1.ChipEXO.R1R2.Paired.Align_R2_minus.sorted.bw
Citation¶
Ye Z, Chen Z, Sunkel B, Frietze S, Huang TH, Wang Q, Jin XV. Genome-wide analysis reveals positional-nucleosome-oriented binding pattern of pioneer factor FOXA1. Nucleic Acids Res 2016 Jul 25. PMID 27458208.